publications
publications by categories in reversed chronological order. generated by jekyll-scholar.
2025
-
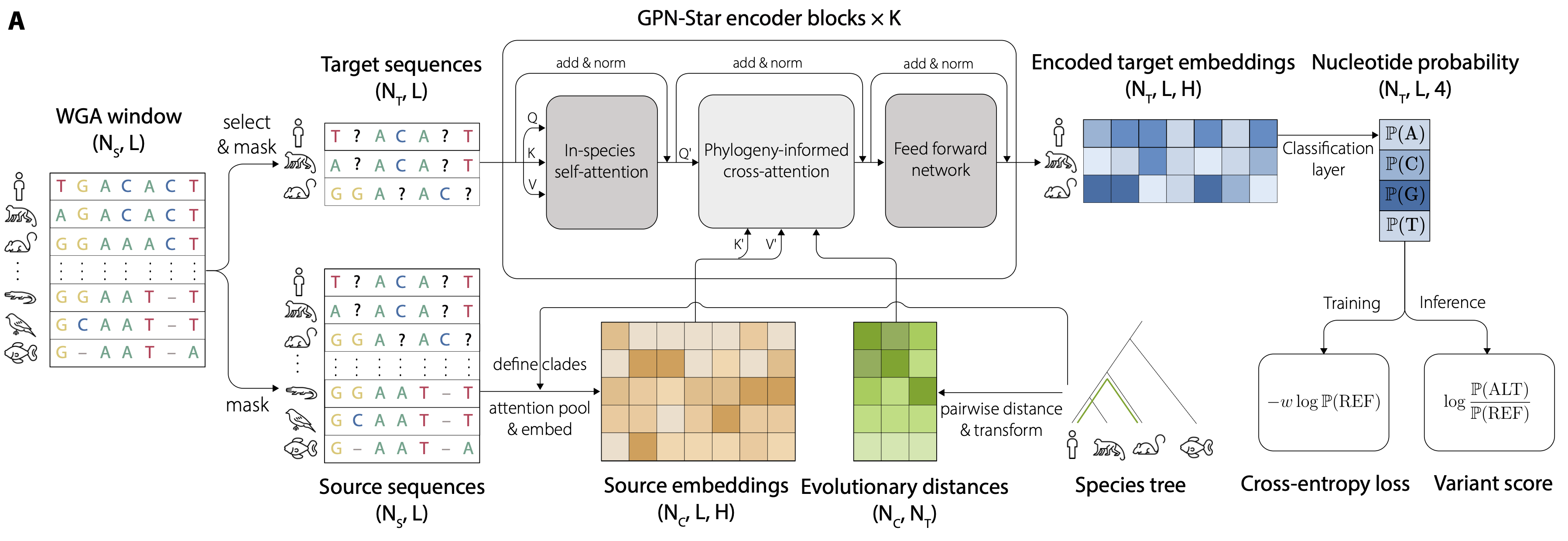 Predicting functional constraints across evolutionary timescales with phylogeny-informed genomic language modelsChengzhong Ye*, Gonzalo Benegas*, Carlos Albors, Jianan Canal Li, Sebastian Prillo, and 3 more authorsbioRxiv, 2025
Predicting functional constraints across evolutionary timescales with phylogeny-informed genomic language modelsChengzhong Ye*, Gonzalo Benegas*, Carlos Albors, Jianan Canal Li, Sebastian Prillo, and 3 more authorsbioRxiv, 2025Genomic language models (gLMs) have emerged as a powerful approach for learning genome-wide functional constraints directly from DNA sequences. However, standard gLMs adapted from natural language processing often require extremely large model sizes and computational resources, yet still fall short of classical evolutionary models in predictive tasks. Here, we introduce GPN-Star (Genomic Pretrained Network with Species Tree and Alignment Representation), a biologically grounded gLM featuring a phylogeny-aware architecture that leverages whole-genome alignments and species trees to model evolutionary relationships explicitly. Trained on alignments spanning vertebrate, mammalian, and primate evolutionary timescales, GPN-Star achieves state-of-the-art performance across a wide range of variant effect prediction tasks in both coding and non-coding regions of the human genome. Analyses across timescales reveal task-dependent advantages of modeling more recent versus deeper evolution. To demonstrate its potential to advance human genetics, we show that GPN-Star substantially outperforms prior methods in prioritizing pathogenic and fine-mapped GWAS variants; yields unprecedented enrichments of complex trait heritability; and improves power in rare variant association testing. Extending beyond humans, we train GPN-Star for five model organisms – Mus musculus, Gallus gallus, Drosophila melanogaster, Caenorhabditis elegans, and Arabidopsis thaliana – demonstrating the robustness and generalizability of the framework. Taken together, these results position GPN-Star as a scalable, powerful, and flexible new tool for genome interpretation, well suited to leverage the growing abundance of comparative genomics data.
-
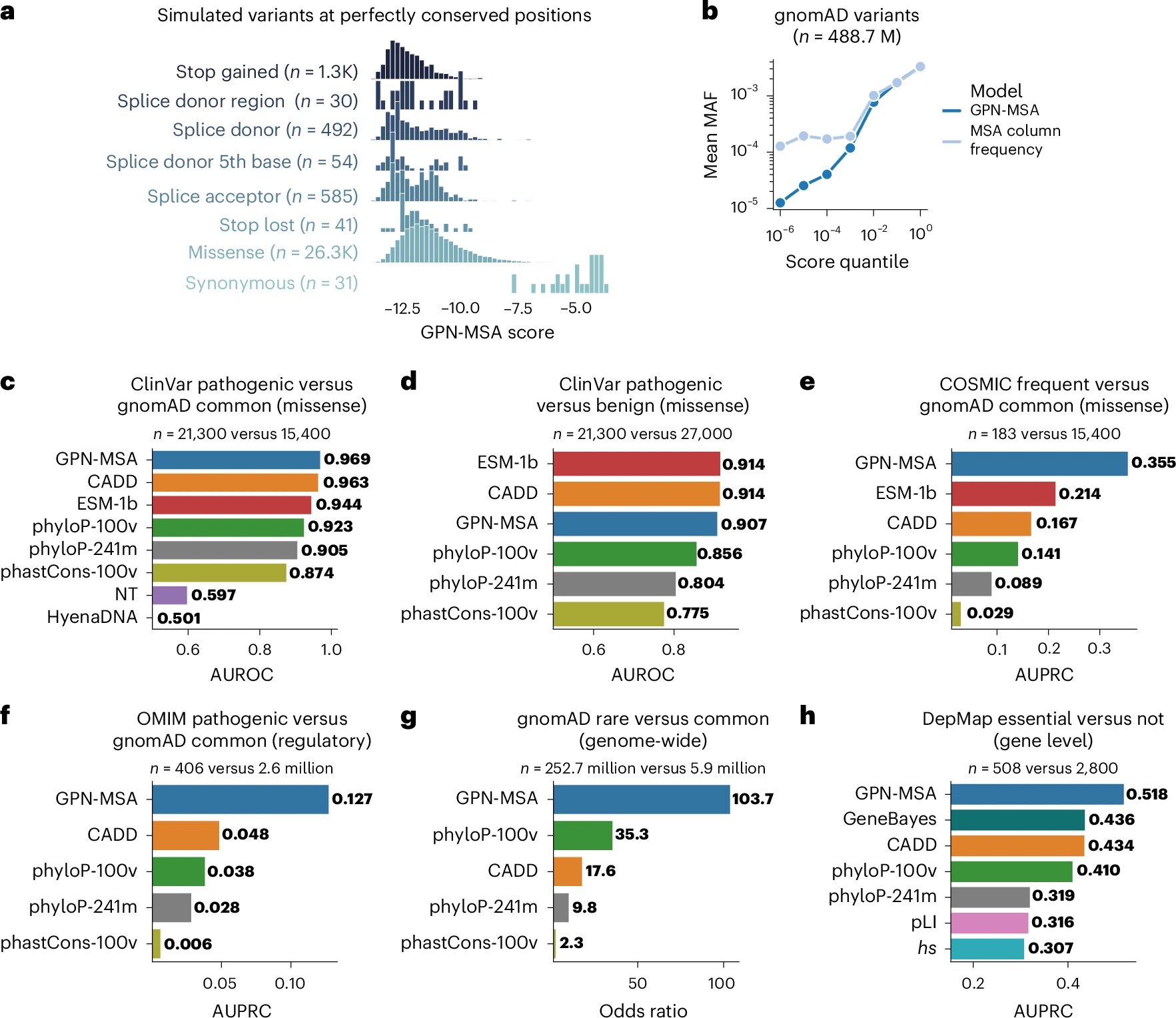 A DNA language model based on multispecies alignment predicts the effects of genome-wide variantsGonzalo Benegas, Carlos Albors, Alan J Aw, Chengzhong Ye, and Yun S SongNature Biotechnology, 2025
A DNA language model based on multispecies alignment predicts the effects of genome-wide variantsGonzalo Benegas, Carlos Albors, Alan J Aw, Chengzhong Ye, and Yun S SongNature Biotechnology, 2025Protein language models have demonstrated remarkable performance in predicting the effects of missense variants but DNA language models have not yet shown a competitive edge for complex genomes such as that of humans. This limitation is particularly evident when dealing with the vast complexity of noncoding regions that comprise approximately 98% of the human genome. To tackle this challenge, we introduce GPN-MSA (genomic pretrained network with multiple-sequence alignment), a framework that leverages whole-genome alignments across multiple species while taking only a few hours to train. Across several benchmarks on clinical databases (ClinVar, COSMIC and OMIM), experimental functional assays (deep mutational scanning and DepMap) and population genomic data (gnomAD), our model for the human genome achieves outstanding performance on deleteriousness prediction for both coding and noncoding variants. We provide precomputed scores for all ~9 billion possible single-nucleotide variants in the human genome. We anticipate that our advances in genome-wide variant effect prediction will enable more accurate rare disease diagnosis and improve rare variant burden testing.
-
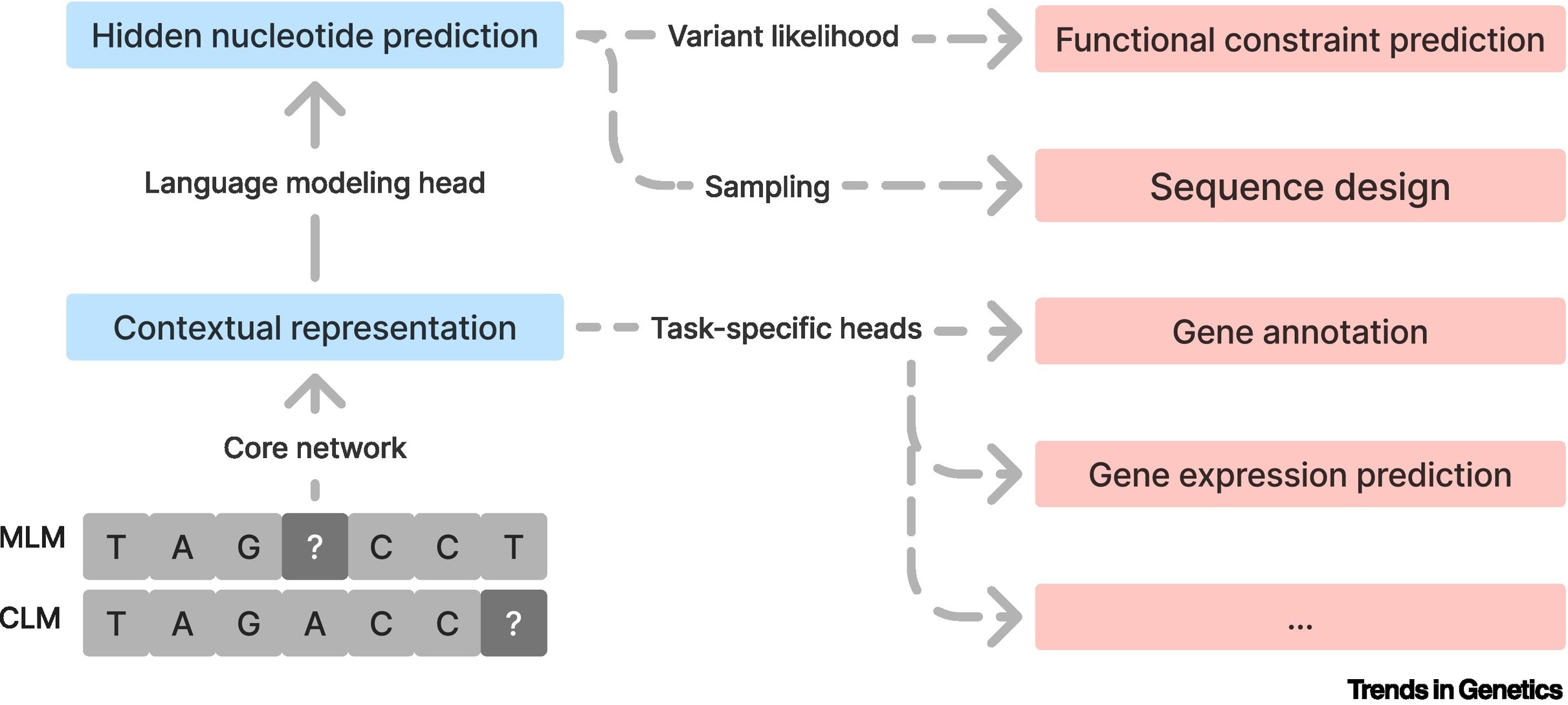 Genomic language models: opportunities and challengesGonzalo Benegas*, Chengzhong Ye*, Carlos Albors*, Jianan Canal Li*, and Yun S SongTrends in Genetics, 2025
Genomic language models: opportunities and challengesGonzalo Benegas*, Chengzhong Ye*, Carlos Albors*, Jianan Canal Li*, and Yun S SongTrends in Genetics, 2025Large language models (LLMs) are having transformative impacts across a wide range of scientific fields, particularly in the biomedical sciences. Just as the goal of natural language processing is to understand sequences of words, a major objective in biology is to understand biological sequences. Genomic language models (gLMs), which are LLMs trained on DNA sequences, have the potential to significantly advance our understanding of genomes and how DNA elements at various scales interact to give rise to complex functions. To showcase this potential, we highlight key applications of gLMs, including functional constraint prediction, sequence design, and transfer learning. Despite notable recent progress, however, developing effective and efficient gLMs presents numerous challenges, especially for species with large, complex genomes. Here, we discuss major considerations for developing and evaluating gLMs.
- Benchmarking DNA Sequence Models for Causal Regulatory Variant Prediction in Human GeneticsGonzalo Benegas, Gökcen Eraslan, and Yun S SongbioRxiv, 2025
- A Phylogenetic Approach to Genomic Language ModelingCarlos Albors, Jianan Canal Li, Gonzalo Benegas, Chengzhong Ye, and Yun S SongIn International Conference on Research in Computational Molecular Biology, 2025



2023
-
 DNA language models are powerful predictors of genome-wide variant effectsGonzalo Benegas, Sanjit Singh Batra, and Yun S. SongProceedings of the National Academy of Sciences, 2023
DNA language models are powerful predictors of genome-wide variant effectsGonzalo Benegas, Sanjit Singh Batra, and Yun S. SongProceedings of the National Academy of Sciences, 2023The expanding catalog of genome-wide association studies (GWAS) provides biological insights across a variety of species, but identifying the causal variants behind these associations remains a significant challenge. Experimental validation is both labor-intensive and costly, highlighting the need for accurate, scalable computational methods to predict the effects of genetic variants across the entire genome. Inspired by recent progress in natural language processing, unsupervised pretraining on large protein sequence databases has proven successful in extracting complex information related to proteins. These models showcase their ability to learn variant effects in coding regions using an unsupervised approach. Expanding on this idea, we here introduce the Genomic Pre-trained Network (GPN), a model designed to learn genome-wide variant effects through unsupervised pretraining on genomic DNA sequences. Our model also successfully learns gene structure and DNA motifs without any supervision. To demonstrate its utility, we train GPN on unaligned reference genomes of Arabidopsis thaliana and seven related species within the Brassicales order and evaluate its ability to predict the functional impact of genetic variants in A. thaliana by utilizing allele frequencies from the 1001 Genomes Project and a comprehensive database of GWAS. Notably, GPN outperforms predictors based on popular conservation scores such as phyloP and phastCons. Our predictions for A. thaliana can be visualized as sequence logos in the UCSC Genome Browser (https://genome.ucsc.edu/s/gbenegas/gpn-arabidopsis). We provide code (https://github.com/songlab-cal/gpn) to train GPN for any given species using its DNA sequence alone, enabling unsupervised prediction of variant effects across the entire genome.
- choros: correction of sequence-based biases for accurate quantification of ribosome profiling dataAmanda Mok, Robert Tunney, Gonzalo Benegas, Edward W. J. Wallace, and Liana F. LareaubioRxiv, 2023
Ribosome profiling quantifies translation genome-wide by sequencing ribosome-protected fragments, or footprints. Its single-codon resolution allows identification of translation regulation, such as ribosome stalls or pauses, on individual genes. However, enzyme preferences during library preparation lead to pervasive sequence artifacts that obscure translation dynamics. Widespread over- and under-representation of ribosome footprints can dominate local footprint densities and skew estimates of elongation rates by up to five fold. To address these biases and uncover true patterns of translation, we present choros, a computational method that models ribosome footprint distributions to provide bias-corrected footprint counts. choros uses negative binomial regression to accurately estimate two sets of parameters: (i) biological contributions from codon-specific translation elongation rates; and (ii) technical contributions from nuclease digestion and ligation efficiencies. We use these parameter estimates to generate bias correction factors that eliminate sequence artifacts. Applying choros to multiple ribosome profiling datasets, we are able to accurately quantify and attenuate ligation biases to provide more faithful measurements of ribosome distribution. We show that a pattern interpreted as pervasive ribosome pausing near the beginning of coding regions is likely to arise from technical biases. Incorporating choros into standard analysis pipelines will improve biological discovery from measurements of translation.Competing Interest StatementThe authors have declared no competing interest.
2022
-
 Robust and annotation-free analysis of alternative splicing across diverse cell types in miceGonzalo Benegas, Jonathan Fischer, and Yun S SongeLife, Mar 2022
Robust and annotation-free analysis of alternative splicing across diverse cell types in miceGonzalo Benegas, Jonathan Fischer, and Yun S SongeLife, Mar 2022Although alternative splicing is a fundamental and pervasive aspect of gene expression in higher eukaryotes, it is often omitted from single-cell studies due to quantification challenges inherent to commonly used short-read sequencing technologies. Here, we undertake the analysis of alternative splicing across numerous diverse murine cell types from two large-scale single-cell datasets—the \textitTabula Muris and BRAIN Initiative Cell Census Network—while accounting for understudied technical artifacts and unannotated events. We find strong and general cell-type-specific alternative splicing, complementary to total gene expression but of similar discriminatory value, and identify a large volume of novel splicing events. We specifically highlight splicing variation across different cell types in primary motor cortex neurons, bone marrow B cells, and various epithelial cells, and we show that the implicated transcripts include many genes which do not display total expression differences. To elucidate the regulation of alternative splicing, we build a custom predictive model based on splicing factor activity, recovering several known interactions while generating new hypotheses, including potential regulatory roles for novel alternative splicing events in critical genes like \textitKhdrbs3 and \textitRbfox1. We make our results available using public interactive browsers to spur further exploration by the community.
